- Share
- Share on Facebook
- Share on LinkedIn
Background
In western countries, new primary glioblastomas (GBM) are diagnosed in 3.2 per 100,000 persons every year (CBTRUS report(1)). Much work is done in medicine, biology, chemistry, mathematics, physics and other fields to improve the prognosis of glioma patients. Modifications of current clinical radiation therapy protocols (hyper- or hypofractionation, hyperoxygenation, etc.) have not provided better results and patients are irradiated to date according to standard protocols that have only been poorly improved for decades (e.g., delivery of 2 Gy daily fractions for 5 to 6 weeks) (2–4). For more than ten years, we are developing a novel form of radiotherapy, known as Microbeam Radiation Therapy (MRT), in which X-rays generated by a synchrotron light source are collimated into a set of of quaisi-parallel microbeams (MBs) of a few tens of microns wide and separated by a few hundred microns (5). The microbeam irradiation geometry allows very high dose (hundreds of grays) deposition in the microbeam paths (peak dose) while tissue slices located in-between microbeams receive only 5-10% of the peak dose (valley dose). Radiation geometry and dose profiles are shown in Figure 1.
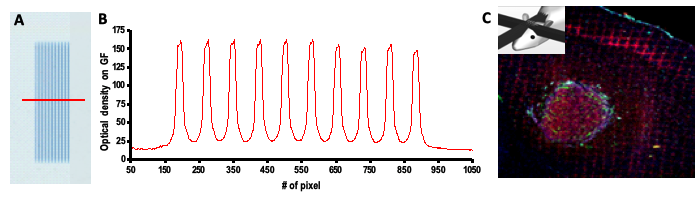
The surprisingly high radiotolerance of normal brain tissue to microbeam exposure is associated with the radioresistance of normal brain vasculature to collimated microbeams (6–8). Conversely, the vasculature in malignant tumours is a promising therapeutic target, because it differs from normal blood vessels morphologically and functionally (9, 10), exhibiting high tortuosity, increased diameters, high permeability and abnormal contractility (11–14). The response of this neo-vasculature to very high radiation doses deposited in the paths of the microbeams drastically differs from that observed in normal, mature blood vessels (15).
Objectives
Investigation of biological processes underlying MRT efficacy
MRT-induced changes to tumour micro-environment
We have successfully tested MRT therapeutic efficacy on the 9L gliosarcoma rat model (6, 16–20) and showed that a microbeam exposure, using only two orthogonal ports, stops the growth of intracerebral rat 9L gliosarcomas, a particularly radio-resistant tumour (21). The median survival time increased by a factor of 3.25, and 80% of tumours were controlled 2 months after exposure (18). We did not observe any change in the morphometric parameters of the vasculature in the contralateral, unidirectionally irradiated hemisphere, while 9LGS exhibited significant changes in tumour blood volume and oxygenation. These observations demonstrated for the first time the role of the vascular component in MRT therapeutic efficacy.
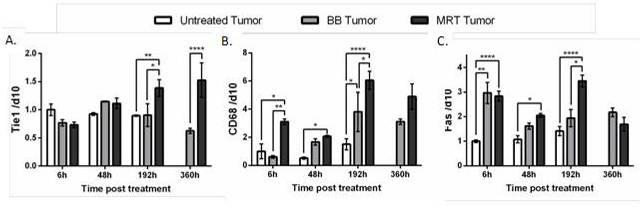
On the 9LGS gliosarcoma model, the comparison of an unidirectional irradiation delivered by MRT or by a synchrotron broad beam irradiation (BB) have clearly demonstrated the better efficacy of MRT on tumour growth control and rats survival (22). The physiological and molecular mechanisms of MRT action are not yet fully elucidated but recent studies have shown specific inflammatory responses and immune levels to MRT (22, 23). Indeed, compared to a BB treatment, MRT induces significantly higher CD68 (monocyte/macrophage marker) and Tie1 endothelial receptor transcripts, which are known to be involved in the attachment of immune cells derived from monocytes (Figure 3). This transcriptomic indication of preferential recruitment of macrophages after MRT is accompanied by a significantly higher level of transcripts related to the induction of cell death (Fas) (22).
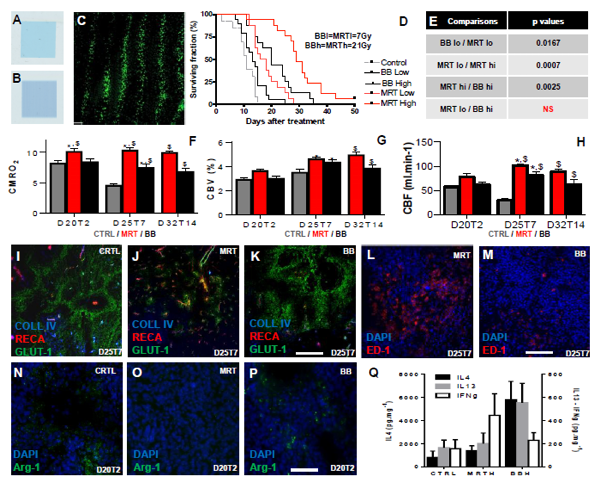
In another experiment recently performed on the F98 glioblastoma model, we were able to demonstrate improved tumour control after MRT compared with conventional RT using a BB generated with a hospital irradiator. We delivered BB at 7 or 21 Gy. In the MRT mode, we applied the same doses to the valleys, and thereby obtained peak doses of 81 and 241 Gy in the microbeam paths, respectively, as determined by Monte Carlo simulations. We showed that MRT led to the same tumour control for a dose 3 times lower than BB, and that the lifespan of animals could be further increased by escalating MRT doses without inducing normal brain toxicity (Figure 4D-E). Preliminary studies performed by Magnetic Resonance Imaging (MRI) revealed that better survival was associated with increased oxygen consumption, tumour blood volume and blood flow (Figure 4F-H). This change resulted in decreasing tumour hypoxia (GLUT-1, Figure 4I-K) showing that MRT, more than BB, changes tumour environment. MRT, which deposits hundreds of Gy, killed more tumour cells, mainly in the microbeam paths which occupied 44% of the total irradiated tumour volume, and attracted significantly more macrophages than tumours irradiated by BB, as shown in Figure 4L-M. Our most important finding from our recent experiment is that macrophages detected in non-irradiated and BB irradiated tumours exhibited the M2 phenotype (Arg-1 positive) whilst no labelling was found in MRT-treated tumours (Figure 4N-P). We have also demonstrated using Bioplex technology that M2-driving cytokines were more frequently expressed in BB-treated tumours, whilst IFN, the main M1-driving cytokine, was found in MRT-exposed tumours (Figure 4Q).
Macrophages, tumour growth and irradiation
The role of the immune system in cancer progression and cancer treatment has gained increasing focus over the last decade. Macrophages are one of the major populations of immunological tumour infiltrating cells and affecting the tumour microenvironnement (24). They respond to environmental cues in a polarized manner, subclassified into M1 (or ‘classically activated') and M2 (or ‘alternatively activated') responses (25, 26)( Figure 5). They may also indirectly promote cytotoxicity by activating other cells of the immune system, such as NK cells and T cells (27). The M1 phenotype is triggered by chemokines such as interferon-gamma (IFN), tumour necrosis factor-alpha (TNFα) or lipopolysaccharide (LPS), granulocyte macrophage-colony stimulating factor (GM-CSF) (28–30), microbial infections and by increased contact and phagocytosis of necrotic cell debris (31, 32). These macrophages display an increased production of nitric oxide (NO) reactive oxygen intermediate (ROI) and reactive nitrogen intermediate (RNI) and promote an anti-tumour response through Interleukin (IL)-12-mediated Th1 activation (33, 34). In contrast, the M2 phenotype results from an alternative activation of macrophages and is mainly present in Tumour Associated Macrophages (TAM). The TAMs are driven to M2 phenotype by several stimuli such as hypoxia, macrophage-colony stimulating factor (M-CSF), IL-4, IL-10, IL-13, IL-21, or Activin-A, as well as corticosteroids, prostaglandins (PGs), vitamin D3, and the chemokine receptor, C–C receptor 4 (CCR4) (35). M2-like polarization macrophages are deemed to promote tumour growth by inhibiting specific immune responses and by stimulating angiogenesis, infiltration and cell proliferation (29, 31, 33, 36). Anti-inflammatory cytokines, including IL-4, IL-10, IL-13, and glucocorticoids polarize macrophages into a M2 phenotype and inhibit macrophage function and activation. M2 production of matrix metallopeptidase 2 (MMP2) also contributes to tumour invasion and metastasis, as well as the secretion of immunosuppressive chemokines (35).
TAM phenotype modulation was recently shown to be efficient in tumour control by reducing growth of gliomas in mice (37). A reversible switch from M1 to M2 phenotype can take only few hours (31); depending on the local environment and is associated with poor prognosis in numerous cancers (mammary carcinoma, Hodgkin’s disease, glioma (38, 39). Macrophages (MΦ ) have been shown to home to malignant tissues after irradiation, mainly through HIF-1 and SDF-1 over-expressions (40). After exposure to 15 Gray dose of ionizing radiation, about 50% of the cells observed in brain tumours were monocytes/macrophages (40). This phenomenon, called the “tumour bed effect”, occurs at doses as low as 5 Gy and is linearly correlated with the radiation dose (41).
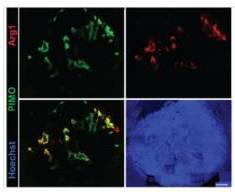
We have shown that irradiation of hypoxic tumour tissues favors the acquisition of M2 macrophage phenotype (19). Beyond cytokine activation, tumour oxygenation is one of the most recognized stimulus in macrophage polarization. Indeed, M1 TAMs are mostly found in the tumour periphery where tissue oxygenation is normal. Conversely, M2 TAMs reside in the hypoxic and necrotic areas at the center of the lesion (42) (Figure 6). Using in vitro studies, we have also demonstrated that primary cultures of non-polarized macrophages (M0) grown in hypoxia express the Arginase 1 and in a manner dependent on the oxygen level since this expression increases strongly at 0.2% oxygen (Figure 7). In an equally interesting way, we have also demonstrated that M1 macrophages grown in hypoxia lose their M1 characteristics in favor of M2 markers. However, we hypothesize that the formation of cellular debris induced by radiotherapies with high necrotic capacity (such as microbeam radiation therapy, please refer above) allows the repolarization of TAMs in M1 (32). Indeed, Barker et al. have demonstrated that macrophage contact and phagocytosis of necrotic but not apoptotic debris increased expression of CD40, M1 marker, antigen presentation function and anti-tumour T lymphocyte activation. The authors concluded that the contact of macrophages with necrotic debris leads to a classical activation, with the development of an inflammatory phenotype M1 (32). According to most recent experiments, which need to be confirmed on larger number of animals, MRT appears to be more efficient than BB at modifying tumour microenvironment, inducing tumour cell death and increasing infiltration by macrophages driven to the M1 phenotype. Therefore, this project is based on the following assumptions:
- Acute cell death induced by high dose MRT produces more cellular debris than irradiation by BB. MRT then increases infiltration by macrophages, facilitates their access to cellular debris which converts macrophages to the M1 phenotype.
- MRT-induced tumour debulking leads to re-oxygenation of the surviving tumour which drives tumour associated macrophages to the M1 phenotype. This phenomenon is intimately related to the differential vascular response between MRT and BB.
- The MRT-induced M1 phenotype can be maintained in vivo and promotes tumour control.
MRT in large animals
In our previous studies, only few long term survivor rats were obtained after a unique dose fraction, mainly because the dose required to kill every tumour cell is not compatible with brain radiotolerance (18). It appears more and more relevant to associate MRT to a conventional treatment, as broad beam (BB) in a fractionated scheme, preparing a dose regimen applicable to future patients in clinical trials. However, the exact patient repositioning required for daily delivery of MRT fractions through identical ports is not possible because of the submillimetre width of the microbeams (50 µm). Therefore, the number of MRT sessions will be limited to the number of different non-colinear ports usable per patient. Until now MRT has been mainly delivered using one or two orthogonal ports (unique session (18, 19, 43)) but MRT might also be applied to brain tumours using a temporal fractionation. First evidences of the technical feasibility and the therapeutic relevance of temporally fractionated MRT have already been given (44). Preliminary data obtained in our laboratory demonstrated that a fraction in the MRT mode, given at the end of a temporally fractionated BB treatment, resulted in a significantly better control of brain tumours in rats than BB only.
In a preliminary study, we evaluated the efficiency of a therapeutic boost delivered by synchrotron MRT on F98 brain tumours in rats after 3 temporally separated BB fractions. We showed that a subsequent microbeam boost induced significant and more prolonged G1 tumour cell arrest than BB exposures alone. MRT-irradiated tumours displayed lower cell density, cell proliferation indices and infiltrative index and more vascular damages compared with BB-irradiated ones. These greater effects of an MRT boost on tumour cells led to a complete stop of tumour growth in vivo during about 4 weeks while BB-treated tumours recurred within few days after the last radiation fraction. These results show for the first time the relevance of MRT as a radiation boost delivered after conventional radiation therapy since additional exposures to microbeams also significantly increased the median survival time (MST) of animals compared with conventional irradiations alone.
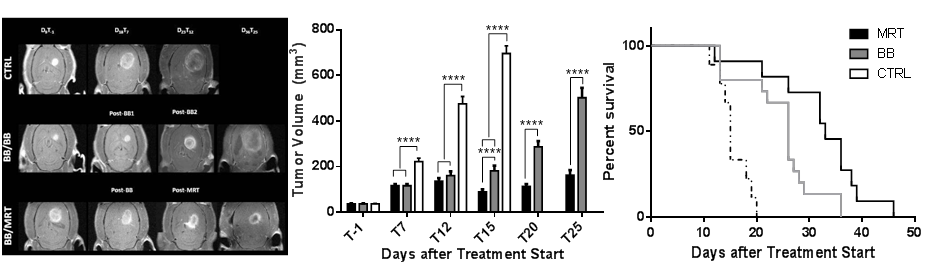
We anticipate that even by delivering only 1/3 of the total radiation dose in the MRT mode, those MRT fractions will be more efficient in treating brain tumours than if the whole radiation dose is applied in the broad beam mode. Thus, we will evaluate the efficiency of MRT delivered before, during or after several BB fractions to palliate brain tumours in rats. Indeed, because MRT shows preferential effects on immature and developing vasculature (18, 45) we assume that MRT boost delivered at treatment start could dramatically reduce tumour perfusion. The following BB fraction efficiency on surviving tumour cells would be then altered, improved by MRT-induced alterations, e.g. in tumour vessels and the microenvironment or reduced if hypoxia would be increased. Alternatively, high MRT doses delivered at the end of the conventional treatment might ablate radio-resistant tumour cell clones remaining after low dose BB fraction exposures. These hypotheses need to be carefully verified and radiation schedules optimized. This project is a new one and stands as one of the last steps towards clinical application of MRT where microbeam radiation therapy would be applied as a relevant and efficient therapeutic boost for glioma management.
We are able from February 2017 to irradiate internal tumours with accurate dosimetry and targeting but MRT irradiations of GBM in vet will start in 2020. We want to profit from the ESRF 2019 shutdown to install and commission the patient positioning system (purchased and set by our long term collaborator ESRF in 2019) in order to anticipate Ministry Agreements and setup validation by French authorities.
As MRT operates at a median energy of 105 keV (half attenuation ~5.5cm), no superficial tumours will be treated and we will directly irradiate deep-seated GBM in dogs using the optimized MRT-Boost regimen. Single threshold doses for normal brain tolerance will be delivered. The irradiation procedure involves external fractionated radiotherapy, that will be done by Pr. Devauchelle and his team. Animals will travel from Paris and their housing ensured by Dr. Devron Gaud in Grenoble (Clinique de l’Esplanade, Grenoble). This vet trial will be designed as a classical phase II/III. We will progressively increase dose and number of MRT fractions in a classical dose escalation 3+3 regimen to probe the dose limitation that could occur with the two RT modalities (6x8Gy regimen). The tumour evolution and normal tissue tolerance will be compared to a large amount of data already obtained in Paris using conventional treatments for similar tumours. Follow up of animals will be ensured by vet doctors and owners. Tissues will be carefully examined at autopsy (agreement from the owner obtained before entering the trial) by our collaborative pathologists in Bern.
We will be able to evaluate efficiencies and radiation induced side effects MRT-boost on gliomas. We will then suggest a safe and efficient approach to move to clinical trials and decide, based on physiological and biological evidences, the optimized way to treat patients using the MRT-Boost protocols.
Clinical transfer and phase I trial setup
During the next 5 years, another main task of STROBE will be to design the road map to clinical trials, to prepare authorization files (ASN, Ministry, etc.) and receive authorizations from countries to perform human clinical trials. At UGA7442, we have experience in preparing and coordinating bio-medical projects in synchrotron research at various scales. We have performed the first human clinical trials using synchrotron X-rays for cardiovascular imaging and brain tumour treatment. The first metastasis-bearing patients were treated in June 2012 and the trial ended last July with Pr J Balosso as PI. Based on these experiences, we expect the clinical transfer of Flash/MRT to be realistic within the frame of this 2019-2024 period. Taking advantage of the acquired experience with SSRT, the initial human candidates for MRT feasibility phase I trial, will be brain oligo-metastases treated with MRT as a boost replacing a rising part of the classical three 11 Gy fractions or of a unique 20 Gy fraction. One then two ports will be used. A progressive replacement of half of the planning dose will be proposed. The schema of 3+3 patients’ steps will be followed. A second phase I/II trial also inspired from SSRT project will be MRT of locally angiogenic relapses of GBM. As it will be re-irradiations, MRT will be used as single approach according to a dose escalation schema. A dose up to a valley dose of 20 supplementary Gy will be delivered in at least 3 fractions using different port each. The optimal dose in MRT could then be tested versus stereotactic X-ray classical re-irradiation as a randomized trial. The next step will be to introduce MRT into initial treatment of GBM and combination with FlashRT and other dose modulation approaches developed in other projects (nanoparticles, targeted pharmaceuticals, etc.). Patients will be able to come from any European country and will all be accepted as in patients at the Grenoble University Hospital.
References
- Dolecek TA, Propp JM, Stroup NE, Kruchko C (2012) CBTRUS statistical report: primary brain and central nervous system tumours diagnosed in the United States in 2005-2009. Neuro Oncol 14 Suppl 5:v1–49.
- Stupp R, et al. (2009) Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 10(5):459–466.
- Stupp R, Roila F (2008) Malignant glioma: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol 19 Suppl 2:ii83–5.
- Stupp R, Brada M, van den Bent MJ, Tonn J-C, Pentheroudakis G (2014) High-grade glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. Available at: http://www.ncbi.nlm.nih.gov/pubmed/24782454 [Accessed July 14, 2014].
- Brauer-Krisch E, et al. (2010) Effects of pulsed, spatially fractionated, microscopic synchrotron X-ray beams on normal and tumoural brain tissue. Mutat Res 704(1-3):160–166.
- Laissue JA, et al. (1998) Neuropathology of ablation of rat gliosarcomas and contiguous brain tissues using a microplanar beam of synchrotron-wiggler-generated X rays. Int J Cancer 78(5):654–660.
- Slatkin DN, Spanne P, Dilmanian FA, Gebbers JO, Laissue JA (1995) Subacute neuropathological effects of microplanar beams of x-rays from a synchrotron wiggler. Proc Natl Acad Sci U S A 92(19):8783–8787.
- Dilmanian FA, et al. (2007) Tissue-sparing effect of x-ray microplanar beams particularly in the CNS: is a bystander effect involved? Exp Hematol35(4 Suppl 1):69–77.
- Jain RK, et al. (2007) Angiogenesis in brain tumours. Nat Rev Neurosci 8(8):610–622.
- Carmeliet P, Jain RK (2000) Angiogenesis in cancer and other diseases. Nature 407(6801):249–257.
- Yancopoulos GD, et al. (2000) Vascular-specific growth factors and blood vessel formation. Nature 407(6801):242–248.
- Jain RK (2005) Normalization of tumour vasculature: an emerging concept in antiangiogenic therapy. Science (80- ) 307(5706):58–62.
- Jain S, et al. (2011) Cell-specific radiosensitization by gold nanoparticles at megavoltage radiation energies. Int J Radiat Oncol Biol Phys79(2):531–9.
- Abdul-Karim MA, Al-Kofahi K, Brown EB, Jain RK, Roysam B (2003) Automated tracing and change analysis of angiogenic vasculature from in vivo multiphoton confocal image time series. Microvasc Res 66(2):113–125.
- Sabatasso S, et al. (2011) Microbeam radiation-induced tissue damage depends on the stage of vascular maturation. Int J Radiat Oncol Biol Phys80(5):1522–1532.
- Bouchet A, et al. (2012) Chalcone JAI-51 improves efficacy of synchrotron microbeam radiation therapy of brain tumours. J Synchrotron Radiat19(Pt 4):478–482.
- Serduc R, et al. (2009) Synchrotron microbeam radiation therapy for rat brain tumour palliation-influence of the microbeam width at constant valley dose. Phys Med Biol 54(21):6711–6724.
- Bouchet A, et al. (2010) Preferential effect of synchrotron microbeam radiation therapy on intracerebral 9L gliosarcoma vascular networks. Int J Rad Onc Biol Phys 78(5):1503–1512.
- Serduc R, et al. (2008) Brain tumour vessel response to synchrotron microbeam radiation therapy: a short-term in vivo study. Phys Med Biol53(13):3609–3622.
- Smilowitz HM, et al. (2006) Synergy of gene-mediated immunoprophylaxis and microbeam radiation therapy for advanced intracerebral rat 9L gliosarcomas. J Neurooncol 78(2):135–143.
- Bencokova Z, et al. (2008) Molecular and cellular response of the most extensively used rodent glioma models to radiation and/or cisplatin. J Neurooncol 86(1):13–21.
- Bouchet A, et al. (2016) Better efficacy of synchrotron spatially micro-fractionated radiotherapy than uniform radiotherapy on glioma. Int J Radiat Oncol.
- Bouchet A, et al. (2013) Early Gene Expression Analysis in 9L Orthotopic Tumour-Bearing Rats Identifies Immune Modulation in Molecular Response to Synchrotron Microbeam Radiation Therapy. PLoS One 8(12):e81874.
- Mantovani A, Germano G, Marchesi F, Locatelli M, Biswas SK (2011) Cancer-promoting tumour-associated macrophages: new vistas and open questions. Eur J Immunol 41(9):2522–5.
- Sica A, Mantovani A (2012) Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122(3):787–95.
- Stout RD, et al. (2005) Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Immunol 175(1):342–9.
- Gabrilovich DI, Ostrand-Rosenberg S, Bronte V (2012) Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12(4):253–68.
- Mantovani A, Sozzani S, Locati M, Allavena P, Sica A (2002) Macrophage polarization: tumour-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23(11):549–55.
- De Palma M, Lewis CE (2013) Macrophage regulation of tumour responses to anticancer therapies. Cancer Cell 23(3):277–86.
- Lewis CE, Pollard JW (2006) Distinct role of macrophages in different tumour microenvironments. Cancer Res 66(2):605–12.
- Biswas SK, Sica A, Lewis CE (2008) Plasticity of macrophage function during tumour progression: regulation by distinct molecular mechanisms. J Immunol 180(4):2011–7.
- Barker RN, Erwig L, Pearce WP, Devine A, Rees AJ (1999) Differential effects of necrotic or apoptotic cell uptake on antigen presentation by macrophages. Pathobiology 67(5-6):302–5.
- Hagemann T, Biswas SK, Lawrence T, Sica A, Lewis CE (2009) Regulation of macrophage function in tumours: the multifaceted role of NF-kappaB. Blood 113(14):3139–46.
- Cook J, Hagemann T (2013) Tumour-associated macrophages and cancer. Curr Opin Pharmacol 13(4):595–601.
- Talmadge JE, Donkor M, Scholar E (2007) Inflammatory cell infiltration of tumours: Jekyll or Hyde. Cancer Metastasis Rev 26(3-4):373–400.
- Lamagna C, Aurrand-Lions M, Imhof BA (2006) Dual role of macrophages in tumour growth and angiogenesis. J Leukoc Biol 80(4):705–13.
- Pyonteck SM, et al. (2012) Deficiency of the macrophage growth factor CSF-1 disrupts pancreatic neuroendocrine tumour development.Oncogene 31(11):1459–67.
- Rossi ML, et al. (1989) The mononuclear cell infiltrate compared with survival in high-grade astrocytomas. Acta Neuropathol 78(2):189–93.
- Nishie A, et al. (1999) Macrophage infiltration and heme oxygenase-1 expression correlate with angiogenesis in human gliomas. Clin Cancer Res5(5):1107–13.
- Kioi M, et al. (2010) Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest 120(3):694–705.
- Milas L, Ito H, Hunter N, Jones S, Peters LJ (1986) Retardation of tumour growth in mice caused by radiation-induced injury of tumour bed stroma: dependency on tumour type. Cancer Res 46(2):723–7.
- Leblond MM, et al. (2015) Hypoxia induces macrophage polarization and re-education toward an M2 phenotype in U87 and U251 glioblastoma models. Oncoimmunology 5(1):e1056442.
- Laissue JA, et al. (1998) Neuropathology of ablation of rat gliosarcomas and contiguous brain tissues using a microplanar beam of synchrotron-wiggler-generated X rays. Int J Cancer 78(5):654–60.
- Serduc R, et al. (2009) First trial of spatial and temporal fractionations of the delivered dose using synchrotron microbeam radiation therapy. J Synchrotron Radiat 16(Pt 4):587–590.
- Bouchet A, et al. (2013) Synchrotron microbeam radiation therapy induces hypoxia in intracerebral gliosarcoma but not in the normal brain. Radiother Oncol 108(1):143–8.
- Share
- Share on Facebook
- Share on LinkedIn